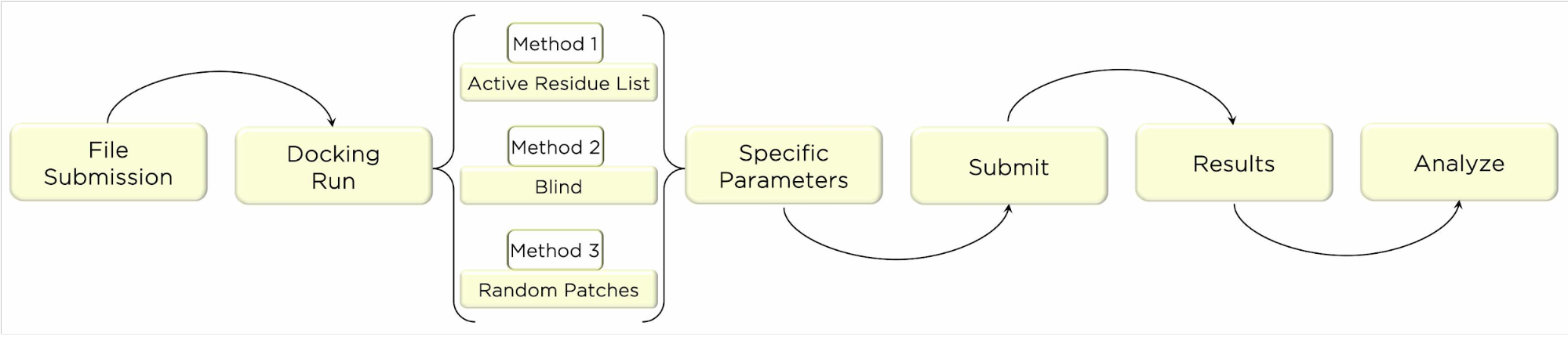
The aim of this protocol is to provide basic knowledge on running the freely available web server HADDOCK (https://bianca.science.uu.nl/haddock2.4/). This web server constitutes a valuable tool using a data-driven docking approach. HADDOCK(van Zundert et al. 2016) gathers important information to define Ambiguous Interaction Restraints (AIRs), or the residues at the interface for each molecule. These can be attained by integrating experimental (NMR chemical shifts, mutagenesis data, etc) and/or predicted information with an empirical forcefield. The result is an energy function accounting for all interactions and from which we want to sample its minima. After identifying the key residues at the interface, HADDOCK calculates and evaluates all distances between the interfacial groups of molecule A and molecule B. This molecular docking procedure brings the molecules together with random interface orientations, for which the final binding score is calculated. The docking process relies on 3 steps (it0, it1 and itw) of gradient driven energy minimization and molecular dynamics simulations. In it0 stage, molecules are rigid and the energy minimization relies strongly on AIRs data. The second stage, it1, introduces flexibility and increases temperature while maintaining restraints on the interface allowing its packing. The final stage, itw, adds in solvation and short molecular dynamics simulations. In the end, a clustering analysis is performed to identify the best binding modes of all the docking solutions from itw stage (van Zundert et al. 2016). (Sample)